Причини та типи атаксій, лабораторні й генетичні дослідження для діагностики спадкових і набутих форм, включаючи метаболічні та імуноопосередковані порушення.
Атаксія є загальним проявом різних неврологічних станів, включаючи інсульт, злоякісні новоутворення головного мозку, розсіяний склероз, травму, токсичний вплив, інфекцію та вроджені дефекти мозочка. Прогресуюча атаксія часто викликає діагностичну невизначеність у практиці невролога і у багатьох випадках залишається недіагностованою (або ідіопатичною).
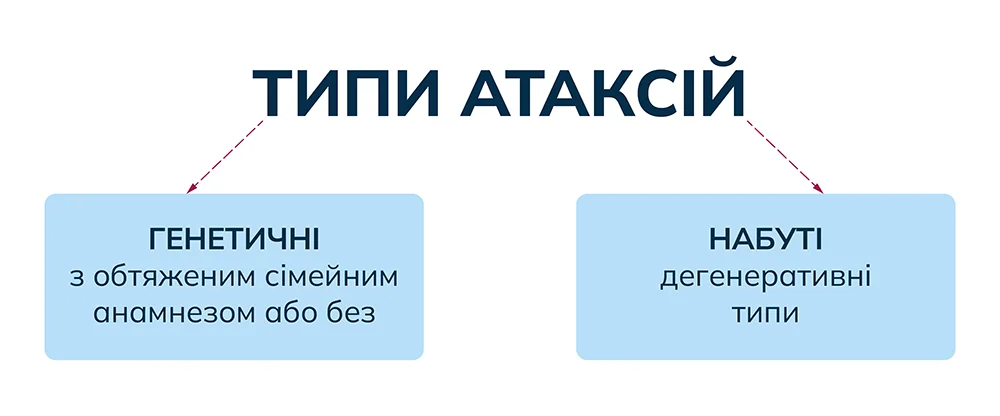
Спадкові (генетичні) атаксії можуть мати аутосомно-домінантний, аутосомно-рецесивний, X-зчеплений або цитоплазматичний типи успадкування.
Метаболічні порушення (наприклад хвороба Німана – Піка типу С, хвороба Тея – Сакса), навіть попри те, що успадковуються, можуть виявлятися атаксією в пізньому віці у пацієнта без обтяженого сімейного анамнезу, що, своєю чергою, підкреслює необхідність ретельного клінічного огляду та лабораторного обстеження.
Набута прогресуюча атаксія може бути:
З метою діагностики атаксій Лабораторія Др. Рьодгера пропонує наступні дослідження:
Наша панель атаксії / спастичної параплегії включає гени, що пов’язані зі спадковими неврологічними розладами, що характеризуються атаксією та спастичною параплегією, включаючи спіноцеребральну атаксію (домінантну та рецесивну). Ці розлади зазвичай мають спільні симптоми, і їх можна чітко диференціювати лише за допомогою молекулярно-генетичного тестування. Традиційно атаксія і спастична параплегія класифікуються в окремі категорії. Однак нещодавно отримана інформація показує, що ці захворювання мають спільні гени, шляхи та механізми, тому наша панель охоплює обидва синдроми та включає спектр захворювань атаксії та спастичності.
Наша панель атаксії / спастичної параплегії є найкращим вибором не лише для пацієнтів, які демонструють дисбаланс ходи та некоординовану ходу, але також для пацієнтів із спастичним порушенням ходи, спастичною слабкістю.
Найпоширеніші форми спадкової атаксії спричинені мутаціями у вигляді збільшення копій триплетів GAA, тому повна версія нашої панелі включає аналіз повторів.
Кількість генів: 482
Умови доставки в лабораторію: в охолодженому вигляді (при tº +2 +8 ºС) до 48 годин.
Умови доставки у Німеччину: на СПЕЦІАЛЬНОМУ ТЕСТ-ПАПЕРІ від Centogene.
Метод: NGS.
Правила підготовки пацієнта: Особливої підготовки не потрібно. Дозволено легкий сніданок.
Ген FXN кодує утворення білка під назвою фратаксин. Цей білок міститься в клітинах по всьому тілу, з найбільшим вмістом у серці, спинному мозку, печінці, підшлунковій залозі та м’язах, які використовуються для довільних рухів (скелетні м’язи). У клітинах фратаксин міститься в структурах, що виробляють енергію, які називаються мітохондріями. Хоча його функція повністю не вивчена, фратаксин, здається, допомагає зібрати кластери молекул заліза та сірки, які є критичними для функціонування багатьох білків, у тому числі тих, які необхідні для виробництва енергії.
Атаксія Фрідрейха є результатом збільшення кількості копій тринуклеотидного повтору GAA в гені FXN. У людей з цим захворюванням сегмент GAA аномально повторюється від 66 до понад 1000 разів. Довжина тринуклеотидного повтору GAA, очевидно, пов’язана з віком, у якому з’являються симптоми атаксії Фрідрейха. Люди з сегментами GAA, які повторюються менш як 300 разів, як правило, мають пізнішу появу симптомів (після 25 років), ніж ті, у кого тринуклеотидні повтори GAA більші.
Більшість людей з атаксією Фрідрейха мають повтор GAA в обох копіях гена FXN. Приблизно 2 відсотки людей з цим захворюванням мають повтор GAA в одній копії гена FXN і інший тип мутації в іншій копії гена. У більшості цих випадків інша мутація змінює один будівельний блок ДНК (нуклеотид) у гені FXN.
Метод: NAA, SEQU, S-Blot, Kapillarelektrophorese
Матеріал: 2-4 мл ЕДТА крові, 2-8 С
Правила підготовки пацієнта: Особливої підготовки не потрібно. Дозволено легкий сніданок.
Хвороба Німана - Піка типу С – являє собою мультисистемний розлад, викликаний накопиченням холестерину та глікосфінголіпідів у головному мозку та інших органах. Отже, у пацієнтів можуть розвинутися сплено-, гепатомегалія, а також атаксія, вертикальний над’ядерний парез погляду, іноді — дистонія, міоклонус, епілепсія та когнітивний дефіцит. Біопсія не завжди дає остаточну відповідь. Генетичне тестування більш надійне. Визначення оксистеролів у плазмі крові та жовчних кислот у сироватці крові може виявитися корисними недорогими інструментами скринінгу.
2770 Генетична панель «Метаболічні порушення» Invitae
2919 Cento Metabolic MOх Centogene
0412 Жовчні кислоти
0364 Антитіла до гліадину IgA до дезамінованих пептидів гліадину
0365 Антитіла до гліадину IgG до дезамінованих пептидів гліадину
1037 Антитіла до трансглютамінази IGG
1038 Антитіла до трансглютамінази IGА
2864 Ендомізіум – АТ (IGA) / AK G. Endomysium
2892 Ендомізіум-АТ (IGG) / Endomysium-AK (IGG)
0305 Вітамін Е (токоферол)
1230 Коензим Q10
У перше 10-річчя розвиваються хронічна діарея та катаракта. Можуть виявляти помітні відкладення холестанолу в сухожиллях. Підтверджується біохімічним аналізом.
2120 Загальний холестерин
2121 Ліпопротеїни високої щільності
2122 Ліпопротеїни низької щільності
2123 Тригліцериди